Monday Morning Effect
Friday evening. While his friends had already met in the Pub on Shattuck Avenue to celebrate a happy hour, UC Berkeley's Ph.D. student Henry Bryndza was still in the Lab. He wanted to finish preparation of his samples so that he could come over on Monday morning to focus on the NMR measurements, not worrying about sample preparations. In order to suppress chemical reactions which could have started in his samples over the weekend, Henry put them in the liquid nitrogen dewar (T=-196℃).
Henry was working in the Laboratory of Robert Bergman, a renowned UC Berkeley professor who has made a significant contribution to the organic and metallorganic chemistry. Bergman and Bryndza were studying Fischer–Tropsch reactions using exemplary Cobalt and Iridium catalysts[1].
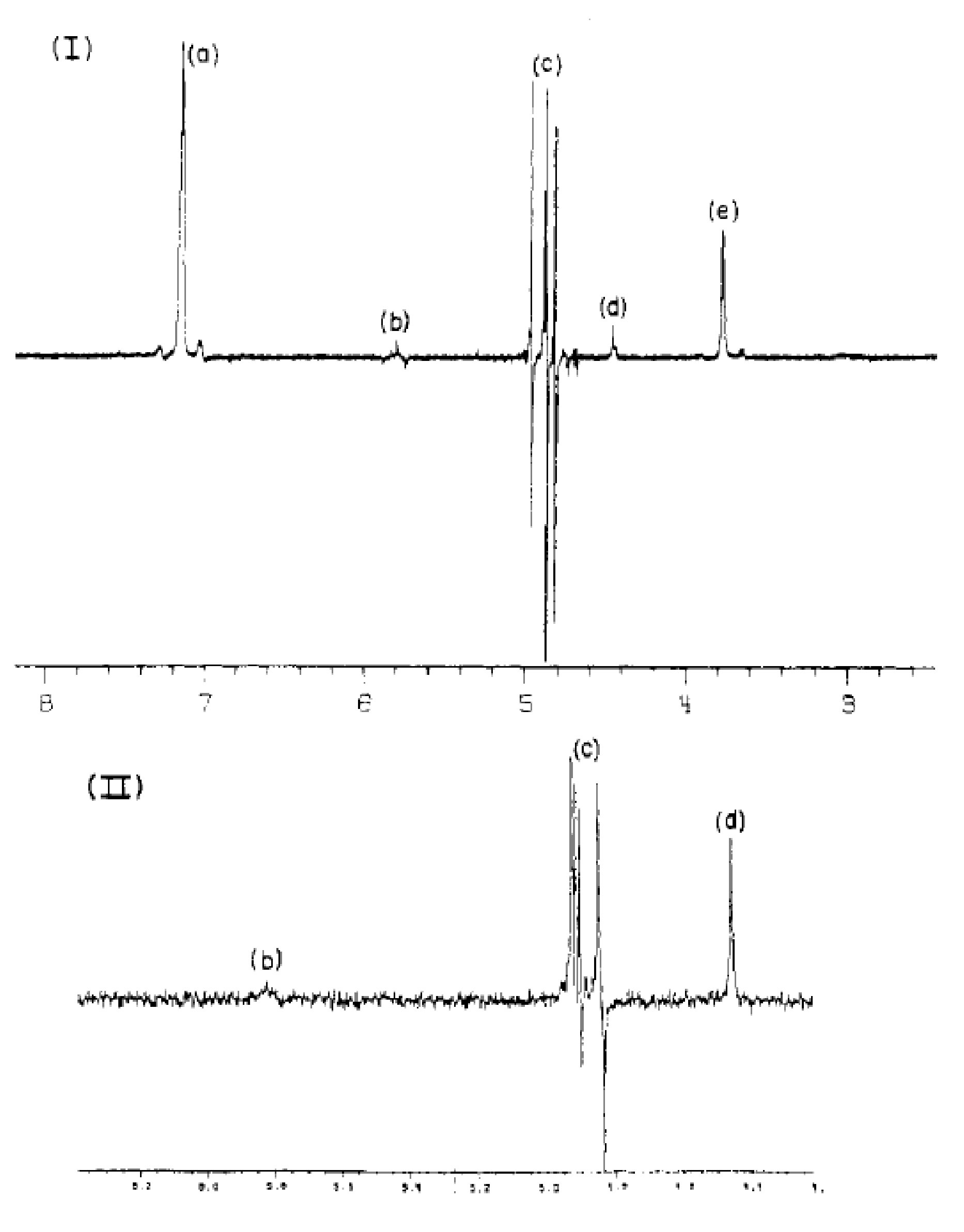
When he came back on Monday, Henry started to observe very interesting phenomena. 1H NMR spectra of the samples he took in the morning showed very weird "negative" NMR peaks (Figure 1). Moreover, the intensity of these peaks decreased day after day during the week when Henry tried to repeat the experiments and completely disappeared by the end of the week[2]. Henry was confused and decided to repeat his measurements. Surprisingly, this phenomenon was not observed every single time but was definitely the strongest on Mondays. Bergman and Bryndza decided to jestingly call this a "Monday phenomenon"; this was the beginning of what was known later as Parahydrogen-Induced Polarization (PHIP).
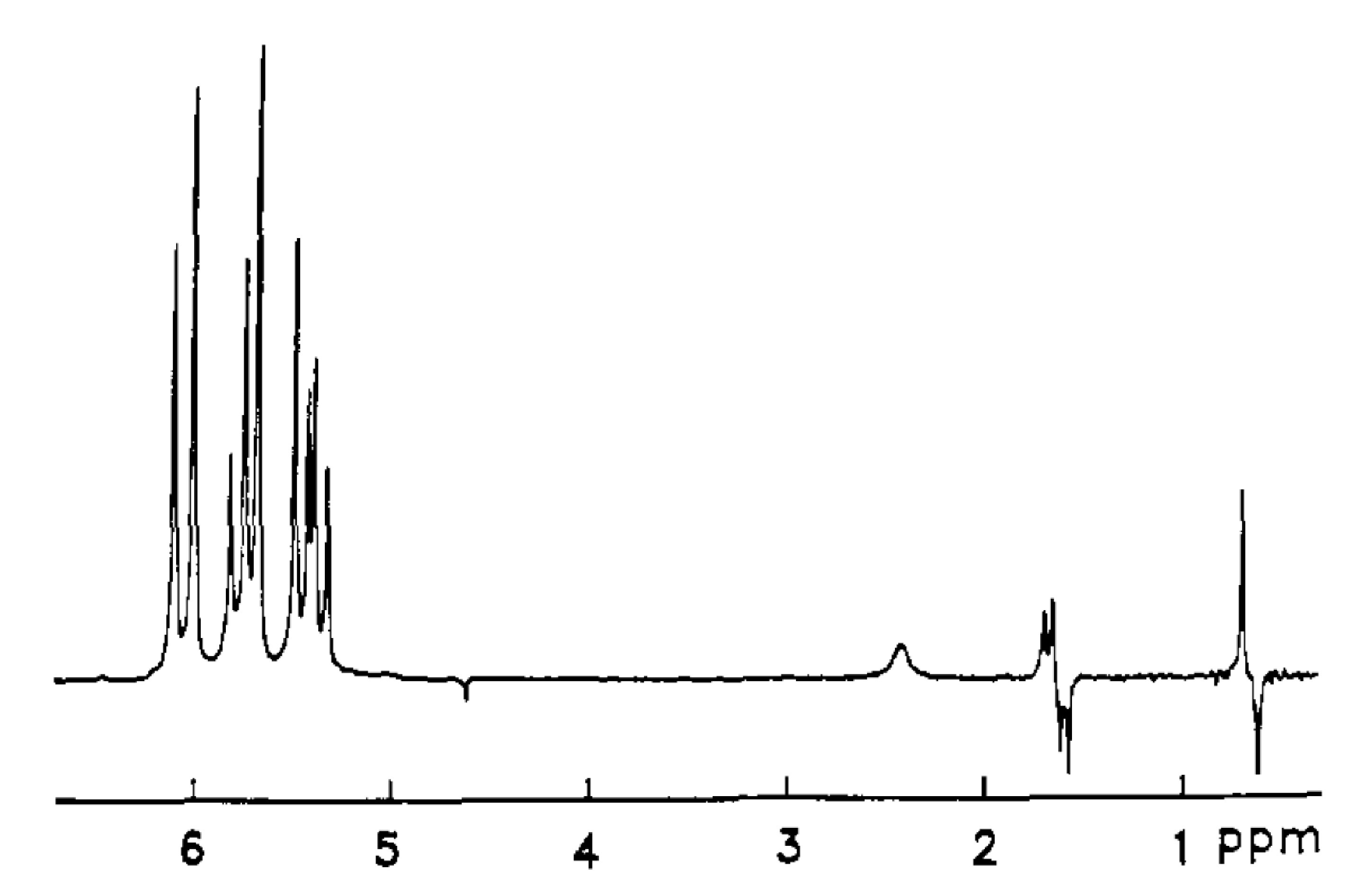
This "Monday morning" puzzle remained unresolved until the International Society of Magnetic Resonance meeting in Rio de Janeiro in June 1986. There, during an evening session, Professor Daniel Weitekamp from Caltech presented his "thought experiment" of using parahydrogen (para-H2) as a source of enhancing NMR signals. The concept and the expected results were immediately published in Physical Review Letters[4]. The experimental demonstration conducted by a Weitekamp's Ph.D. student Russ Bowers followed in July, and brilliantly supported all theoretical predictions (Figure 2)[5].
Now let's talk about physical principles of this effect. As we discussed before, due to the absence of a net nuclear magnetic moment, para-H2 itself does not produce an NMR signal. However, this single nuclear spin state implies that, in a sense, it is cold. Indeed, a comparable degree of spin ordering is obtainable at equilibrium only at temperatures of a few mK and magnetic fields of several Tesla[7]. The brilliance of Wetekamp's idea was to introduce magnetic inequivalence to release this potential signal by connecting the singlet to the triplet states. This would require chemistry, but simple bond cleavage would not suffice. A singlet state of two protons is a relationship of one spin relative to the other and this order would be dissipated if the pair were split and mixed with an ensemble of other such products. Rather, it is necessary that the pair have a special relationship even after being distinguished by magnetic inequivalence. This is called a "pairwise" hydrogen addition and can be realized in hydrogenation reactions in the presence of homogeneous catalysts. To see how it works, let's take as an example the simplest situation and imagine that a chemical reaction leads to the association of para-H2 with a molecule not containing magnetic nuclei.
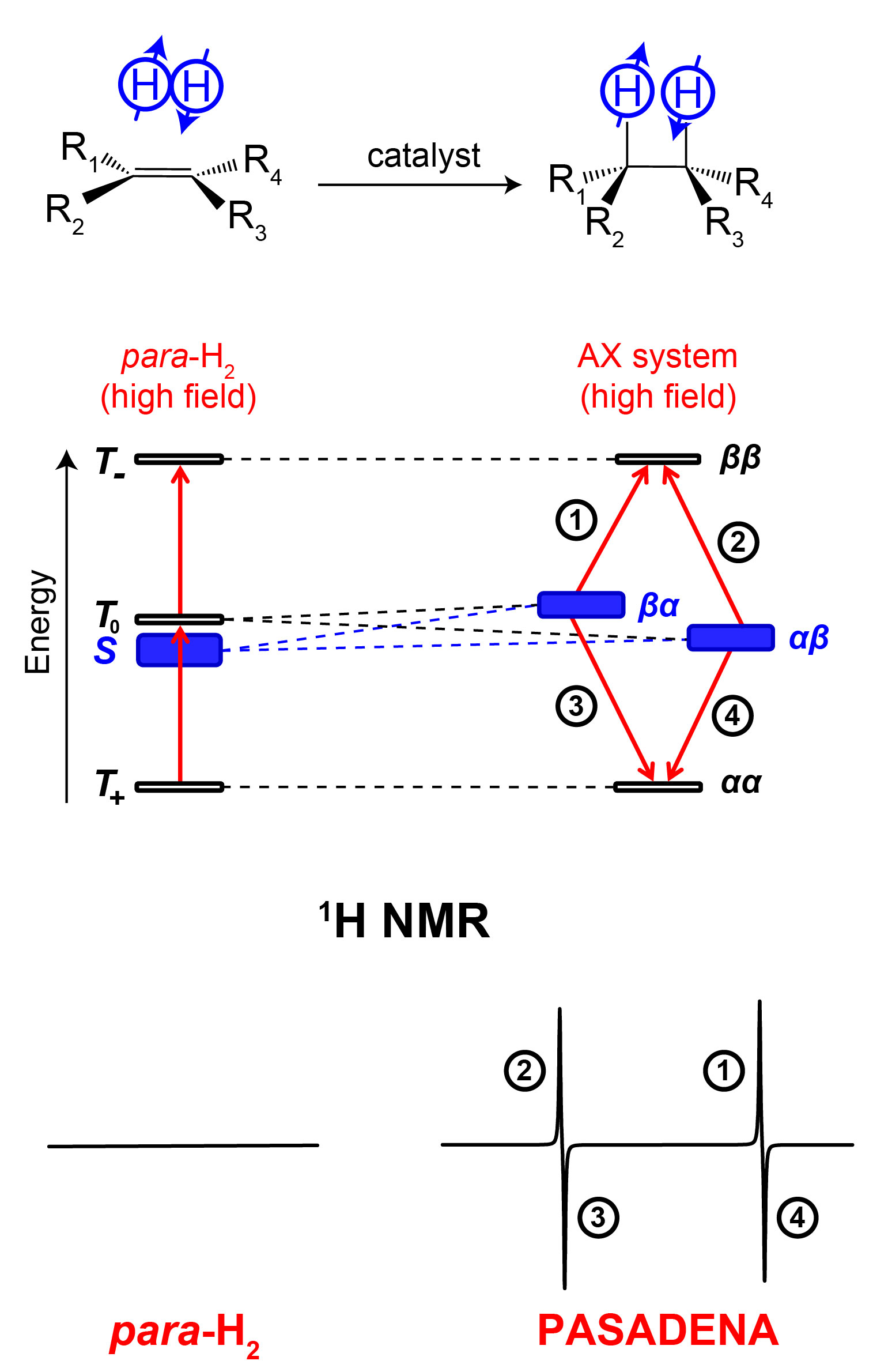
The two-spin system of the hydrogen molecule gives rise to four nuclear spin energy levels. As we described before, three of these energy levels correspond to orthohydrogen, the state with total nuclear spin 1 (triplet state), whereas the remaining fourth energy level corresponds to parahydrogen (singlet state), the state with zero total nuclear spin (Figure 3). Transitions between singlet and triplet spin states are forbidden by symmetry and the spin 0 parahydrogen is NMR-silent.
Now, the incorporation of para-H2 into an asymmetric molecule breaks the symmetry of the singlet spin state. For simplicity, I will consider only the PASADENA experiment, the case where hydrogenation reaction is performed at a high magnetic field (wherein the chemical shift difference between the two para-H2-nascent protons is much greater than the spin-spin coupling J between them). In this situation, the population of the singlet spin state αβ-βα (numerical factor is omitted) of para-H2 is immediately transferred to the population of spin states αβ and βα of the formed spin system.
This can be understood as follows. Because of the chemical reaction, two H atoms from para-H2 suddenly end up in a different molecular environment. This leads to a collapse of the nuclear spin wavefunction αβ-βα into one of the two states, αβ or βα, each with 50% probability. Next, it is easy to deduce from the energy level diagram that the NMR spectrum of the produced in such a manner molecule will contain four peaks grouped in two antiphase multiplets (Figure 3), exactly what was observed in the experiments of Bryndza (Figure 1) and Bowers (Figure 2). The key requirement is that both hydrogen protons from the para-H2 molecule are added together without significant competition from exchange reactions. This is a property of many, but not all, hydrogenations.
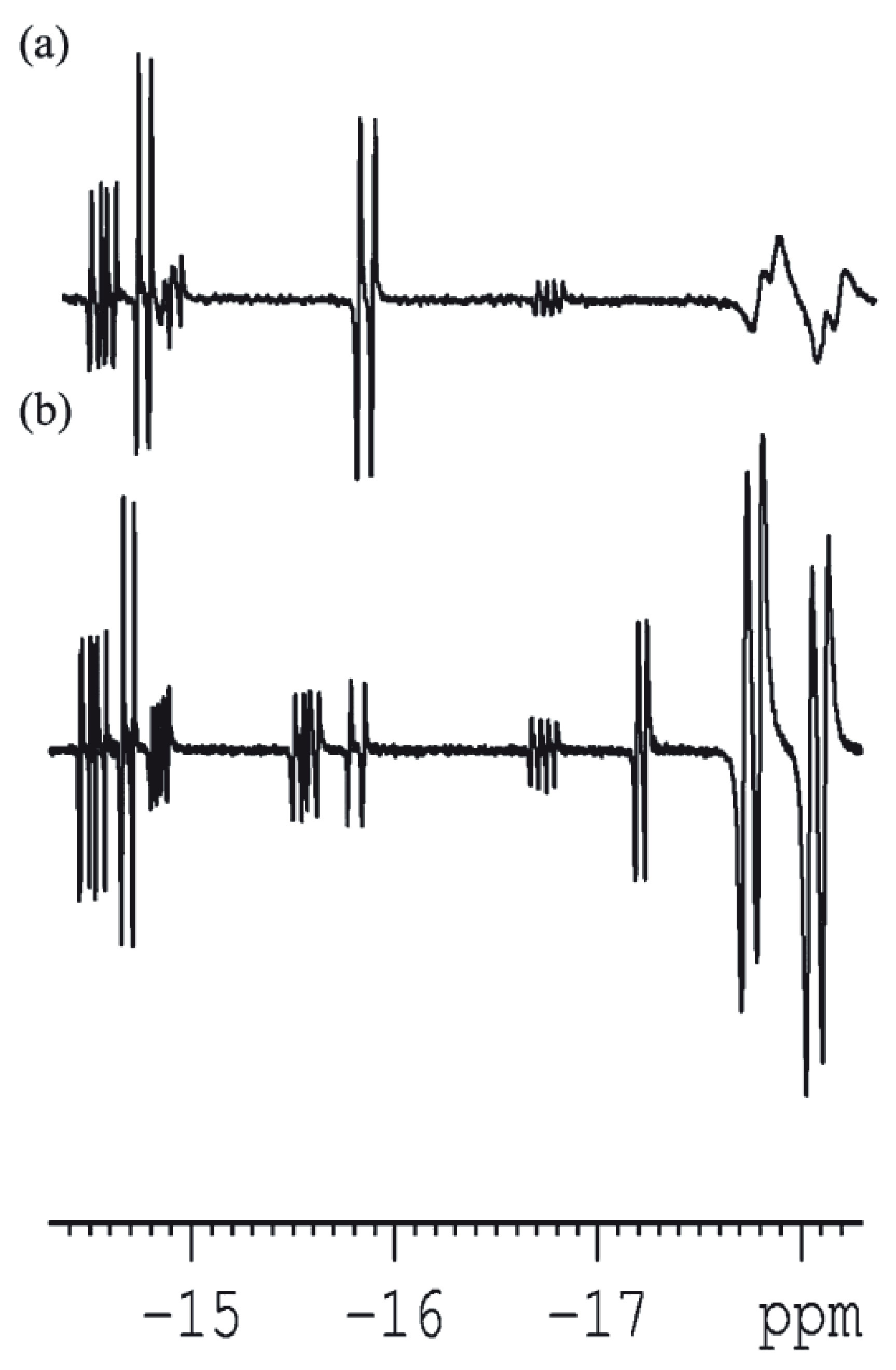
The assignment of the peaks to particular transitions depends on the sign of the J-coupling between the para-H2-nascent hydrogens. When J-coupling is positive, PASADENA multiplets are positive-negative; if J-coupling is negative, the spectral appearance is opposite. This feature is very useful for studying hydrogenation reaction intermediates. Normally, organic molecules possess positive J-couplings between protons; and J-couplings between them are negative in case of metal hydrides. Therefore, in a complex reaction involving many intermediates, it becomes possible to distinguish low-concentration hydrides (possessing negative-positive multiplets) from organic reaction products (Figure 4).
It is also important to realize that PHIP can lead to 100% nuclear spin polarization of the reaction product. In the case of PASADENA experiment, 100% population of para-H2 is split into just two energy levels, making transitions from these levels enhanced by orders of magnitude compared to the thermal case. Theoretically, if all para-H2 molecules are transferred to products in a pairwise manner and relaxation loses are minimized, the reaction product can acquire 100% spin polarization. This would, of course, require an additional step to transfer spin order from αβ and βα into the state αα but this can be readily realized using a simple RF pulse sequence.
Enormous NMR signal enhancements and unique spectroscopic signatures made PHIP a very useful tool in chemistry for more than 25 years to elucidate hydrogenation reaction mechanisms, study metalorganic hydride complexes, and catalysis [6]. However, PHIP can be also used in a very different context. Imagine a suitable molecular precursor which can become a naturally occurring metabolite after hydrogenation. This metabolite can be produced in seconds, with a very high level of nuclear polarization, injected into a living organism and a metabolism of that organism can be monitored by magnetic resonance spectroscopy (MRS) and magnetic resonance imaging (MRI). Today PHIP, and its sister technology SABRE (Signal Amplification By Reversible Exchange) allow to efficiently hyperpolarize dozens of biologically relevant molecules on nuclei such as 1H, 13C, 15N, 19F, 29Si, 31P, 119Sn etc. But this is a story for a separate blog post! :)
It is important to emphasize that only the connection between nuclear spin and rotational degrees of freedom allows this unique situation to take place. Indeed, the fact that the nuclear spin state can be overpopulated simply by cooling is a remarkable quality inherent only to the small hydrogen molecule. Indeed, even though other molecules can have the similar connection between rotational and nuclear spin states (N2, F2 etc.), larger moments of inertia will make overpopulating these states much more challenging task (because of the lower temperature requirements). Moreover, it is very challenging to keep these molecules in the gas state at low temperatures, and the simple rule of making a total wavefunction be a product of individual wavefunctions will no longer hold true. So, it is more likely that hydrogen molecule is the only example when the rules of spin statistics and Pauli's principle can lead to the nuclear spin hyperpolarization.
What excites me about this story is how a purely thought experiment, on the one hand, and a weird experimental phenomenon, on the other hand, emerged into a new discipline and a remarkable tool to study chemical reactions. Moreover, more exciting applications of the para-H2-based hyperpolarization techniques are expected to emerge in biomedicine. I really wish there were more Monday morning effects in science! Who knows but maybe someone today has come to a lab to look at a weird result which will form a new field of study tomorrow.
References[1] J. Bargon. Chance Discoveries of Hyperpolarization Phenomena. eMagRes, 2007.
[2] Private conversations with Robert Bergman and Alex Pines.
[3] P. F. Seidler, H. E. Bryndza, J. E. Frommer, L. S. Stuhi, R. G. Bergman. Synthesis of Trinuclear Alkylidyne Complexes from Dinuclear Alkyne Complexes and Metal Hydrides. CIDNP Evidence for Vinyl Radical Intermediates In the Hydrogenolysis of These Clusters. Organometallics, 1983, 2 (11), 1701-1705.
[4] C. R. Bowers, D. P. Weitekamp. Transformation of Symmetrization Order to Nuclear-Spin Magnetization by Chemical Reaction and Nuclear Magnetic Resonance. Phys. Rev. Lett., 1986, 57 (21), 2645-2648.
[5] C. R. Bowers, D. P. Weitekamp. Parahydrogen and Synthesis Allow Dramatically Enhanced Nuclear Alignment. J. Am. Chem. Soc., 1987, 109 (18), 5541-5542.
[6] J. Natterer, J. Bargon. Parahydrogen-Induced Polarization. Prog. Nucl. Magn. Reson. Spect. 1997, 31, 293-315.
[7] D. Weitekamp. Sensitivity Enhancement Through Spin Statistics. Encyclopedia of Magnetic Resonance, 2007.
[8] S. Colebrooke, S. Duckett, J. Lohman, R. Eisenberg. Hydrogenation studies involving halobis(phosphine)-rhodium(I) dimers: Use of parahydrogen-induced polarisation to detect species present at low concentration. Chem. Eur. J., 2004, 10, 2459–2474.